|
1 Ludwig Institute for Cancer Research, Brussels Branch, 74 avenue Hippocrate UCL 7459, B-1200 Brussels, Belgium and Cellular Genetics Unit, University of Louvain, 74 avenue Hippocrate UCL 7459, B-1200 Brussels. The existence of specific tumor rejection antigens was first demonstrated with chemically induced mouse sarcomas: each tumor was found to express a different antigen [1 ]. Similar findings were made with ultraviolet-induced tumors [2]. Later, the generality of the existence of tumor rejection antigens was questioned when spontaneous mouse tumors were found to be completely incapable of eliciting an immune rejection response [3]. However, further experiments demonstrated that even these tumors express weak transplantation antigens that are potential targets for immune rejection by the syngeneic host [4]. But what is the molecular nature of tumor rejection antigens? And what is the relation between their appearance and the tumoral transformation process? These questions are still unanswered because these antigens, which elicit strong T -cell mediated immune responses, do not stimulate B cells to produce antibodies. It has therefore been impossible to isolate the antigenic molecules by immunoprecipitation. Recently, we have developed a gene transfection approach aimed at identifying directly the genes that code for this type of antigen. It was applied to "tum- " transplantation antigens, which arise on mouse tumor cells when they are treated with mutagenic agents, and to a tumor rejection antigen present on mouse mastocytoma P 815.
In vitro mutagen treatment of mouse tumor cells generates at high
frequency stable immunogenic variants that are rejected by syngeneic
mice [5]. Because of their failure to form tumors, these variants
were named "turn-" as opposed to the original "turn + " cell, which
produces progressive tumors. This phenomenon has been observed on
a large number of mouse tumor cell lines of various types [6]. Most
turn variants express new transplantation antigens not found on
the original turn+ cell. The existence of these tum- antigens was
first demonstrated by transplantation experiments [7] We have studied
a series of tumvariants derived from mastocytoma P815, a tumor induced
in a DBA/2 mouse with methylcholanthrene. From clonal turn + line
P 1, we obtained more than 30 different tum- variants, which rarely
produced progressive tumors even when they were injected at very
large doses. When restimulated in vitro, spleen cells of mice that
had rejected these variants produced cytolytic T cells (CTL) that
lysed preferentially the immunizing turn variant [8]. From these
lymphocytes, we were able to isolate stable CTL clones [9]. Some
of these appeared to be directed against a tumor rejection antigen
of P815: they lysed P1 and all P815-derived cells but not syngeneic
control tumors. Others lysed the immunizing tum- variant, but neither
the original turn+ cell nor the other tum- variants. They therefore
defined new tum- antigens specific for each variant (Fig. 1 ). These
antigens displayed considerable diversity: no antigen was found
twice among 15 tumvariants that were analyzed. By in vitro immunoselection
with anti-tum- CTL clones it was possible to demonstrate that some
tum- variants carry several turn antigens [10]. These experiments
also demonstrated that the tum- antigens defined by CTL are relevant
to the rejection of the variants, as shown by the correlation between
the loss of these antigens and the reversal of the tum- phenotype
[10, 11]. To find an explanation that could reconcile the remarkably
high frequency of tum- variants with their stability and to understand
the source of their diversity, it appeared essential to identify
the antigenic molecules. We failed in our attempts to obtain antibodies
directed against tum- antigens. Therefore, we undertook to clone
directly the relevant genes on the basis of their ability to produce
the antigens recognized by the anti-tum- CTL. 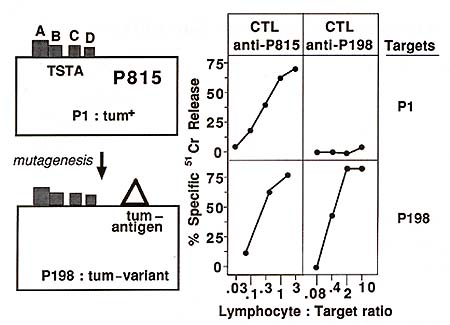
The procedure that we developed for the cloning of the gene coding
for tumantigen P91 A is based on gene transfection. It involves
the use of a highly transfectable P 815 cell line called P 1. HTR
[12] and the detection of antigenexpressing transfectants by their
ability to stimulate CTLs [13]. By transfecting Pl. HTR cells with
a cosmid library prepared with the DNA of a cell expressing turn
antigen P91 A, we obtained transfectants expressing this antigen
at a frequency of 1 per 28 000 [14]. By direct encapsidation of
the DNA of these transfectants into lambda phage heads, we obtained
a cosmid capable of transferring the expression of the antigen.
An 800-bp restriction fragment from this cosmid was found to transfer
the expression of the antigen. This fragment was then used to identify
cosmids containing either the normal or the antigenic allele of
the entire P91A gene as well as complementary (c)DNA clones of the
homologous messenger RNA. The procedure that led to the isolation
of tum- gene P91 A was applied with success to the cloning of tum-
genes P35B and P198, which encode antigens expressed by other tum-
variants derived from P815 [15, 16]. 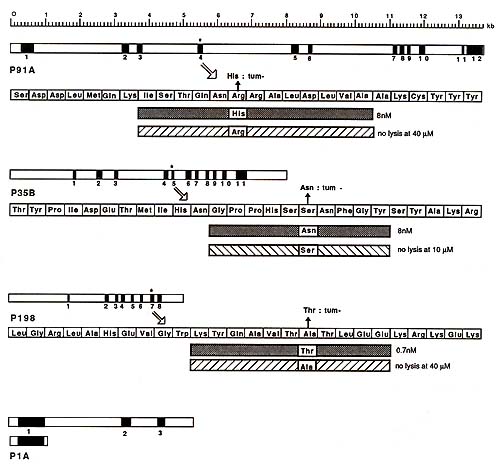
Northern blots probed with the 800-bp fragment of gene P91 A revealed a single messenger RNA species of 2.2 kb. The band was of equal intensity for tum variant P91 and for P1, which does not express the antigen. The expression of antigen P91 A is therefore not due to the activation of a silent gene. The structure of gene P91 A is shown in Fig. 2. It comprises 12 exons spread over 14 kb [17]. It does not show any similarity with Ig, T cell receptor or MHC genes. The complete sequence was obtained. It is unrelated to any sequence presently recorded in the main data banks. A sequence comparison of the normal and tum- alleles of gene P91 A indicated that they differ by a point mutation in the exon which is present in the transfecting 800-bp fragment (Fig. 2). This tum- mutation is a G to A transition that changes an arginine into a histidine in the main open reading frame of the gene [14]. This mutation appears to be the only difference distinguishing the normal from the antigenic allele. The study of the tum- alleles of genes P35B and P198 also revealed that they differ from the normal alleles by a point mutation in an exon (Fig. 2). The general structures and the sequences of the three tum- genes isolated so far are completely unrelated.
The main open reading frame of gene P 91 A encodes a protein of 60 kDa, which does not have atypical N-terminal signal sequence [17]. In vitro translation experiments suggest that the two potential Nglycosylation sites present in the sequence are unused (Godelaine, Amar-Costesec, De Plaen, Beaufay, unpublished results). Antigen P91 A is therefore unlikely to be borne by a membrane protein. This is however hardly surprising, considering the recent demonstration that CTL can recognize influenza antigens corresponding to endogenous proteins remaining inside the cell and considering the observation that CTL recognize small pep tides that bind to surface class I MHC molecules [18-20]. On the basis of this evidence, we examined whether we could also identify a small peptide that would trigger the lysis of P815 cells by antiP91 A CTL. In our search for this peptide we were guided by the location of the tum- mutation. A short peptide (Fig. 2) corresponding to the mutant sequence induced the lysis of P 1 by anti-P91 A CTL. Transfection and peptide studies with H-2k fibroblasts, which expressed also either Kd, Dd or Ld, demonstrated that antigen P91 A is associative with Ld. Antigenic peptides corresponding to the sequence surrounding the turn -mutation were also obtained for genes P35B and P198. They associate with Dd and Kd respectively. Studies with P91 A peptides enabled us to understand the role of the turn mutation. A priori, the mutation could influence either the production of the antigenic peptide or its ability to associate with the Ld molecule (i.e., the aggretope) or also the epitope presented to T cells by the peptide-MHC complex. Having the antigenic P91 A peptide, we prepared the homologous peptide corresponding to the normal allele of the gene. This normal peptide did not induce lysis by anti-P91A CTL, nor did it compete with the mutant peptide. Moreover, we found that the mutant peptide competed effectively to prevent a cytomegalovirusderived peptide from inducing lysis by CTL directed against a Ld-associative cytomegalovirus antigen. The normal peptide did not compete [17] and we concluded that it does not bind to Ld. This indicates that the P 91 A turn mutation generates the aggretope of the antigen, but does not exclude that it also influences the epitope. For antigen P 198, the effect of the mutation appears to be different: here anew epitope is introduced on a normal peptide that is already capable of binding to the Kd presenting molecule.
We have applied the same cloning procedure to the isolation of
the gene coding for a tumor rejection antigen expressed by tumor
P815 [21]. As opposed to the tum- antigens, these antigens are present
on all P815 cells, whether they are mutagenized or not. The study
of antigen-loss variants enabled us to identify four distinct antigens
recognized by different syngeneic CTL clones. They were called P1A,
B, C, and D (Fig.1) [22]. Antigens P 1 A and P 1 B thus defined
in vitro are relevant in vivo, because P815 tumor cells that progressed
in mice after nearly complete initial rejection were found to have
lost the expression of one or both these antigens. Antigens P1 A
and P1 B showed linkage, since several antigenloss variants for
P1A were found to have lost P1 B concurrently. For the transfection
of antigen P1A, we used as recipient cell a P 1 A- B antigen-loss
variant selected from line P 1. HTR with an anti-P1 A CTL clone.
Transfectants expressing both antigens P1A and P1B were obtained
with the genomic DNA ofP1. HTR. This confirmed the close link between
these two antigens. Transfectants were then obtained with a cosmid
library made with the DNA of a genomic transfectant. By directly
packaging the DNA of one of these cosmid transfectants, we obtained
a cosmid that was able to transfect both antigens P 1 A and P 1
B. The structure and the complete sequence of gene P 1 A were then
obtained (Fig. 2). They proved completely different from those of
the tum- genes and of any known gene reported in data banks. Transfection
studies in H2-k fibroblasts previously transfected with either Kd,
Dd, or Ld demonstrated that both P 1 A and P 1 B were presented
to the CTL by the Ld molecule. We compared the sequence of this
gene, cloned from tumor cells, to the sequence of the equivalent
gene cloned from normal cells of the same mouse strain. From a genomic
library made with the DNA of normal DBA/2 mouse kidney we isolated
the gene homologous to gene P 1 A. The analysis of this gene revealed
that its sequence was identical to the sequence of the tumoral gene.
To confirm this, we transfected this normal gene and found that
it transferred the expression of antigens P 1 A and P 1 B as efficiently
as the gene cloned from P815 cells (Fig. 3). The antigenicity is
therefore not the result of a mutation in the tumoral gene, and
P 1 A and P 1 B are presumably two different peptides derived from
the same protein. The tumor specificity of antigens P 1 A and P
1 B can nevertheless be partially explained by the pattern of expression
of the gene. Northern blot analysis revealed 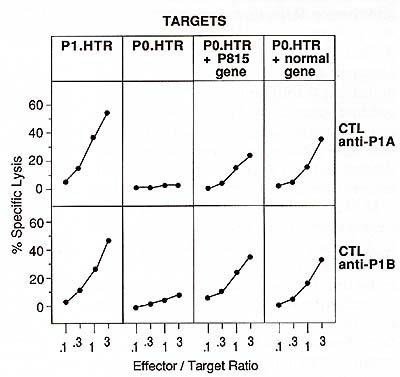
A first conclusion based on the results obtained with the tum- antigens is that mutations throughout all the mammalian genome generate at high efficiency antigenic peptides recognized by T cells, and that this mechanism could account for the presence of specific tumor rejection antigens on carcinogen-induced tumors. However, the study of antigen P 1 A clearly showed that gene P 1 A is identical to its normal counterpart. The apparent tumor specificity of antigen P 1 A seems to be due to a specific regulation of the transcription of the gene rather than to a mutation generating an antigenic peptide. We now have to understand how the immune system may be sensitized against normal peptides to which is should be tolerant, and this without generating an obvious autoimmune pathology. Several hypotheses can be suggested. If the gene encodes an embryonic or oncofetal protein, then the antigen might have disappeared before the establishment of tolerance. If it codes for a differentiation or activation antigen, we can imagine that it is expressed very transiently by a small number of cells, so that tolerance does not develop and that an immune reaction directed against this antigen does not impair normal differentiation or activation. Lastly, if tolerance is actually present for P 1 A, then it must have been broken, and the simultaneous presence on the P815 tumor of other antigens like C and D may be important in that respect. These antigens could indeed be the result of a mutation and therefore be strongly immunogenic like tum antigens. They could possibly trigger an immune response that would facilitate a response against P1A [4]. References 1. Prehn RT, Main JM (1957) Immunity to methylcholanthrene-induced sarcomas. INCI18:769 2. Kripke MI (1981) Immunologic mechanism in UV radiation carcinogenesis. Adv Cancer Res 34: 69 3. Hewitt HB, Blake ER, Walder AS (1976) A critique of the evidence for active host defense against cancer based on personal studies of 27 murine tumors of spontaneous origin. Br J Cancer 33: 241 4. Van Pel A, Vessiere F, Boon T (1983) Protection against two spontaneous mouse leukemias conferred by immunogenic variants obtained by mutagenesis. J Exp Med 157:1992 5. Boon T, Kellerman O ( 1977) Rejection by syngeneic mice of cell variants obtained by mutagenesis of a malignant teratocarcinoma cell line. Proc Natl Acad Sci USA 74:272 6. Frost P, Kerbel R, Bauer E, TartamellaBiondo R, Cefalu W (1983) Mutagen treatments as a means for selecting immunogenic variants from otherwise poorly immunogenic malignant murine tumors. Cancer Res 43: 125 7. Boon T, van Pel A (1978) Teratocarcinoma cell variants rejected by syngeneic mice: protection of mice immunized with these variants against other variants and against the original malignant cell line. Proc Natl Acad Sci USA 75:1519 8. Boon T, van Snick 1, van Pel A, Uyttenhove C, Marchand M (1980) Immunogenic variants obtained by mutagenesis of mouse mastocytoma P815. T lymphocytemediated cytolysis. 1 Exp Med 152: 1184 9. Maryanski lL, van Snick 1, Cerottini lC, Boon T (1982) Immunogenic variants obtained by mutagenesis of mouse mastocytoma P815. Clonal analysis of the syngeneic cytolytic T lymphocyte response. Eur 1 Immuno112:401 10. Maryanski lL, Boon T (1982) Immunogenic variants obtained by mutagenesis of mouse mastocytoma P815. Analysis of variants-specific antigens by selection of antigen-loss variants with cytolytic T -cell clones. Eur 1 Immuno112:406 11. Maryanski 1, Marchand M, Uyttenhove C, Boon T (1983) Immunogenic variants obtained by mutagenesis of mouse mastocytoma P815. Occasional escape from host rejection due to antigen-loss secondary variants. Int 1 Cancer 31: 119 12. Van Pel A, de Plaen E, Boon T (1985) Selection of highly transfectable variant from mouse mastocytoma P815. Somatic Cell Mol Genet 11 :467 13. Wolfel T, van Pel A, de Plaen E, Lurquin C, Maryanski lL, Boon T (1987) Immunogenic (turn-) variants obtained by mutagenesis of mouse mastocytoma P815. Detection of stable transfectants expressing a tum- antigen with a cytolytic T cell stimulation assay. Immunogenetics 26: 178 14. De Plaen E, Lurquin C, van Pel A, Mariame B, Szikora lP, Wolfel T, Sibille C, Chomez P, Boon T (1988) Immunogenic (turn -) variants of mouse tumor P815: cloning of the gene of turn antigen P91 A and identification of the tum- mutation. Proc Natl Acad Sci USA 85:2274 15. Szikora lP, van Pel A, Brichard V, Andre M, van Baren N, Henry P, de Plaen E, Boon T (1990) Structure of the gene of tum- transplantation antigen P 35 B: pre sence of a point mutation in the antigenic allele. EMBO 19:1041 16. Sibille C, Chomez P, Wildmann C, van Pel A, de Plaen E, Maryanski lL, de Bergeyck V, Boon T (1990) Structure of the gene of tum- transplantation antigen P198: a point mutation generates anew antigenic peptide. 1 Exp Med 172: 35 17. Lurquin C, van Pel A, Mariame B, de Plaen E, Szikora lP, lanssens C, Reddehase Ml, Lejeune 1, Boon T (1989) Structure of the gene of tum- transplantation antigen P91 A: the mutated exon encodes a peptide recognized with Ld by cytolytic T cells. Cell 58: 293 18. Townsend ARM, aotch FM, Davey 1 (1985) Cytotoxic T cells recognize fragments of the influenza nucleoprotein. Cell 42:457 19. T ownsend ARM, Roth bard 1, aotch FM, Bahadur a, Wraith D, McMichael Al (1986) The epitopes of influenza nucleoprotein recognized by cytotoxic T lymphocytes can be defined with short synthetic peptides. Cell 44: 959 20. Townsend ARM, Bastin 1, aould K, Brownlee aa (1986) Cytotoxic T lymphocytes recognize influenza haemagglutinin that lacks a signal sequence. Nature 324:575 21. Van den Eynde B, Lethe B, van Pel A, de Plaen E, Boon T (1991) The gene coding for the main tumor rejection antigen of mouse tumor P 815 is identical to the normal gene of the syngeneic DBA/2 mice. 1 Exp Med 173:1373 22. Uyttenhove C, Maryanski 1, Boon T (1983) Escape of mouse mastocytoma P815 after nearly complete rejection is due to antigen-loss variants rather than immunosuppression. 1 Exp Med 157: 1040 23. Hültner L, Moeller 1, Schmitt E, lager a, Reisbach a, Ring 1, Dormer P (1989) Thiol-sensitive mast cell lines derived from mouse bone marrow respond to a mast cell growth-enhancing activity different from both IL-3 and IL-4. 1 Immuno1142:3440 |