|
1 Melbourne Tumour Biology Branch, Ludwig Institute
for Cancer Research, Post Office, Royal Melbourne Hospital, Melbourne,
Vic., 3050 Australia
It has become apparent from studies in vitro that the survival, proliferation and differentiation of progenitor cells and functional activation of the various mature cell types of the haemopoietic system are controlled by a group of glycoprotein regulators, notably erythropoietin, T cell growth factor (IL2) and the colony-stim ulating factors (CSFs) [1, 2]. Four CSFs with distinct biochemical and biological properties have been purified: M-CSF is a selective proliferative stimulus for macrophages [3]; G-CSF for granulocytes [4]; GM-CSF for both granulocytes and macrophages [5]; while multi-CSF (also known as interleukin-3 [6], burst-promoting activity [7], p cell-stimulating factor [8], mast cell growth factor [9] and haemopoietic cell growth factor [10]) stimulates the proliferation not only of neutrophilic granulocytes and macrophages, but also eosinophils, megakaryocytes, erythroid and mast cells. GM-CSF synthesized by mouse lung tissue is a glycoprotein of molecular weight 23000 [5] and is required continuously for the in vitro proliferation of progenitor cells of granulocytes and macrophages, controls the irreversible commitment of these progenitors to the formation of mature granulocytes and macrophages [II] and regulates the functional activity of the mature end cells. Although it has been possible to purify all four CSFs, detailed analysis of many aspects of the biology and biochemistry of these factors has been hampered by the limited quantities available. This problem can be largely circumvented by molecular cloning of the corresponding gene sequences and by using the cloned gene sequence to direct the synthesis of the corresponding factor. We have previously isolated cDNA clones containing partial copies of the GM-CSF mRNA from mouse lung [12]. In this paper we report the isolation of a cDNA clone which contains all of the information required to direct the synthesis of biologically active GM-CSF in simian cas cells [ 13 ].
I. Cloning of Murine GM-CSF cDNA Sequences 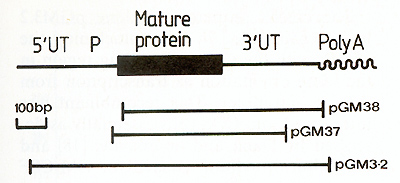 Fig. I. Map of the murine GM-CSF mRNA. The region of the mRNA encoding the mature protein is shown as a thick line, the untranslated regions are designated by UT and the putative precursor peptide by P. The regions contained within clones pGM37, pGM38 and pGM3.2 are indicated with bars  Fig.2. Predicted amino acid sequence of ill urine GM-CSF. The sequence presented is of the mature protein and is that predicted by nucleotide sequence analysis [ 12] of clones pGM37 and pGM38. Owing to a nucleotide sequence difference between the two clones, residue 116 could be either glycine or serine [12] this mRNA indicated that the 3' untranslated region of the mRNA is 319 nucleotides in length and the region encoding the mature protein is 354 nucleotides, leaving some 350 nucleotides for the putative NH2 terminal signal peptide and the 5' untranslated region [12]. The amino acid sequence of GM-CSF deduced from the nucleotide sequence of the mRNA is given in Fig. 2, starting with the first amino acid of the mature protein [ 14]. The protein is predicted to be 118 amino acids in length, with a molecular weight of 13500. The cDNA sequence in clone pGM37 extends about 20 nucleotides 5' to the region encoding the mature protein, into the region encoding the signal peptide, but does not extend to the translational initiation codon. As a prelude to the direct expression of the cloned GM-CSF gene sequence in cell culture, we needed to isolate a cDNA clone containing the entire coding region of the GM-CSF mRNA, including the translation al initiation codon. In order to isolate more GM-CSF cDNA clones we have made use of a cloned T lymphocyte line, LB3 [ 15], in which the synthesis of high levels of G MCSF mRNA is inducible by concanavalin A (see Fig.2 in reference [ 12]). We estimate the abundance of GM-CSF mRNA in conA-stimulated LB3 RNA to be at least two orders of magnitude greater than in lung RNA. We therefore constructed a library of cDNA clones complementary to con-Astimulated LB3 RNA, and screened this library for GM-CSF clones by colony hybridization using a fragment of DNA from pGM38 as a probe. Of 24 GM-CSF cDNA clones purified and examined, one (pGM3.2) appears to contain a substantial, if not complete, copy of the GM-CSF mRNA. The region of GM-CSF mRNA contained in this clone, determined by mapping the location of various restriction endonuclease sites, is illustrated in Fig. I.
In order to be able to express eukaryotic cDNA sequences in cell
culture, we have constructed a vector (pJL) which utilizes the late
promoter of the simian virus SV40 to transcribe inserted DNA sequences
(Fig.3). The vector contains the ß-lactamase gene and origin of
DNA replication from the bacterial plasmid pAT153 [16], the SV40
origin of DNA replication and T antigen coding sequences [ 17] and
a "multicloning site" adjacent to the SV40 late promoter. This multicloning
site contains cleavage sites for several restriction endon ucleases
suitable for insertion of foreign DNA sequences: EcoRI, BamHI, Sacl,
XbaI, Sail and Sma I. When introduced into cultured simian cells,
such as CVl or COS [ 13], this vector is able to replicate and to
transcribe any DNA sequence inserted at the multicloning site. Provided
that translational start and stop codons are included, the inserted
sequence will be translated. 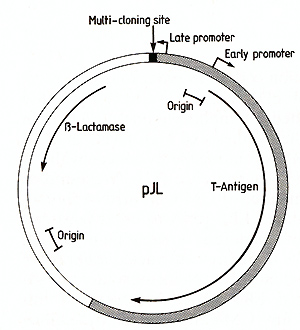 Fig.3. Map of the expression vector pJL. The region derived
from pATl53 [16] is indicated by an open segment,
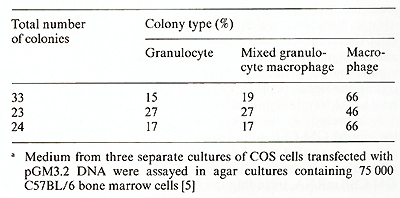 The cDNA sequence of clone pGM3.2 has been installed in the multicloning site of pJL with the G M-CSF coding region in the same orientation as transcription from the late promoter. This recombinant was introduced into COS cells essentially as described by Dana and Sompayrac [ 18] and the culture medium assayed for GM-CSF activity 48 h after transfection. As negative controls, COS cells were also transfected with pJL DNA alone or with two different recombinant plasmids containing incomplete copies of the GM-CSF mRNA; cas cells which received no DNA were also assayed for GM-CSF activity. The various conditioned media were assayed for GM-CSF activity using both the FD cell line, which is absolutely dependent upon the presence of either GM-CSF or multiCSF for growth (A. Hapel, personal communications) and the 32D CL3 cell line [19], which is responsive only to multi-CSF. As shown in Fig. 4, the medi um for cas cells transfected with pGM3.2 DNA was able to support the growth of FD cells whereas media from all of the negative controls were completely inactive. As expected, the medium from pGM3.2-transfected COS cells was inactive on 32D CL3 cells (not shown), indicating that the active factor was GM-CSF rather than multi-CSF. The medium from pGM3.2-transfected COS cells also stimulated the formation of morphologically identifiable granulocyte and macrophage colonies in agar cultures containing bone marrow cells (Table I) [5], the ratio of the three colony types being characteristic of GM-CSF at these concentrations [20]. The full range of biological ac tivities of the factor specified by this cloned gene is currently being assessed. 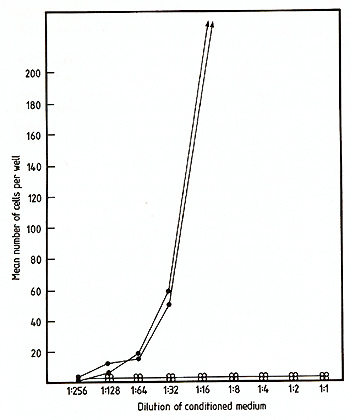
The molecular cloning of genes encoding the various haemopoietic growth regulators should allow the resolution of many outstanding questions and also bring to light hitherto unappreciated problems. By nucleotide sequence analysis of cloned gene sequences, the complete amino acid sequences of murine GM-CSF [12] and multi-CSF [21,22] have been deduced -a goal that was essentially unattainable by conventional biochemical approaches. Comparison of these two sequences has now raised further intriguing issues. As we have previously discussed [12], neither the primary amino acid sequences nor the predicted secondary structures of these two factors show any significant homology, despite the similar activities that they display in stimulating the growth of granulocyte and macrophage colonies from committed progenitor cells. It should now be possible to study in more detail the interaction between these two factors and their receptors, since we can now produce both GM-CSF (this paper) and multi-CSF (N. M. Gough, unpublished work) in a clonally pure form upon introduction of the cloned gene sequences into cas cells. Moreover, by in vitro mutagenesis and expression of the mutated gene sequences it should be possible to dissect the active site (or sites) of the molecules and to ask whether their various activities are determined by the same or different active sites.
The work at the Walter and Eliza Hall Institute was supported
by grants from the Carden Fellowship Fund
1. MetcalfD, Moore MAS (1971) Haemopoietic cells. North Holland, Amsterdam 2. Metcalf D ( 198] ) In: Baserga R ( ed) Tissue growth factors. Springer, New York, pp 343 -384 3. Stanley E. Heard PM (1977) J Biol Chem 252:4305-4312 4. Nicola NA, Metcalf D, Matsumoto M, John son GR (1983) J Biol Chem 258:9017-9021 5. Burgess A W , Camakaris J, Metcalf D ( 1977) J Biol Chem 252: 1998-2003 6. Ihle JN et al. (1982) J Immunol 129: 2431-2436 7. Iscove NN, Roitsch CA, Williams N, Guilbert LJ ( 1982) J Cell Physiol [Suppl] 1: 65- 78 8. Clark-Lewis I, Schrader JW (1981) J Im munol127:1941-1947 9. Yung, Y-P, Eger R, Tertlan G, Moore MAS (1981) J Immunol 127: 794- 799 10. Bazill GW, Haynes M, Garland J, Dexter TM (1983) Biochem J 210: 747- 759 11. Metcalf D, Burgess A W ( 1982 ) J Cell Physiol 111:275-283 12. Gluzman Y (1981) Cel123: 175-182 13. GoughNMetal.(1984)Nature 309:763-767 14. Sparrow Let al. (1985) Proc Natl Acad Sci USA 82:292-296 15. Kelso A, MacDonald HR, Smith KA, Cerottini J-C, Brunner KT (1984) J Immunol 132: 2932-2938 16. Twigg AJ, Sherratt D (1980) Nature 283: 216-218 17. Tooze J ( 1980) Molecular biology of tumour viruses, part 2: DNA tumour viruses. Cold Spring Harbor Laboratory 18. Danna KJ, Sompayrac LM (1982) J Virol Methods 5: 335-341 19. Greenberger JS et al. (1982) In: Baum SJ, Ledney GD, Thierfelder S (eds) Experimental haematology today 1982. Karger, Basel, pp 195-209 20. Burgess AW, MetcalfD (1977) In: Baum SJ, Ledney GD (eds) Experimental haematology today 1977. Springer, New York, pp 135-146 21. Fung MC et al. (1984) Nature 307:233-237 22. Yakota T et al. (1984) Proc Natl Acad Sci USA 81: 1070-1074 |